Let me tell you about my stupidest R mistake.
In the R package that I write, R/qtl, one of the main file formats is a comma-delimited file, where the blank cells in the second row are important, as they distinguish the initial phenotype columns from the genetic marker columns.
I’d gotten some reports that if there were many phenotypes, the import of such a file could take an extremely long time. I ignored the problem (as it wasn’t a problem for me), but eventually it did become a problem for me, and when I investigated, I found the following code.
# determine number of phenotypes based on initial blanks in row 2
n <- ncol(data)
temp <- rep(FALSE,n)
for(i in 1:n) {
temp[i] <- all(data[2,1:i]=="")
if(!temp[i]) break
}
if(!any(temp)) # no phenotypes!
stop("You must include at least one phenotype (e.g., an index).")
n.phe <- max((1:n)[temp])
Here data is the input matrix, and I use a for loop over columns, looking for the first cell for which all preceding cells were empty. If you can understand the code above, I’m sure you’ll agree that it is really stupid. I think the code was in the package for at least five years, possibly even eight.
For a file with 200 individuals and 1500 phenotypes, it would take about 60 seconds to load; after the fix (below), it took about 2 seconds. I spent 58 seconds looking for the first non-blank cell in the second row!
In April, 2009, I fixed it (see the commit at the github repository) by replacing the above with the following.
if(data[2,1] != "")
stop("You must include at least one phenotype (e.g., an index).")
n.phe <- min(which(data[2,] != ""))-1
If you don’t quite understand what I’m talking about, here’s a picture of one of these comma-delimited files; this one has three phenotypes.
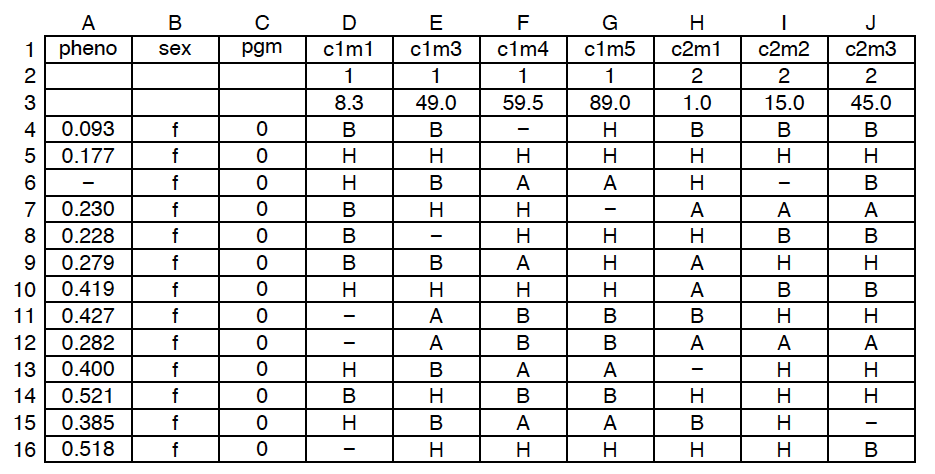
Open source means everyone can see my stupid mistakes. Version control means everyone can see every stupid mistake I’ve ever made.